research areas
|
We use X-chromosome inactivation (XCI) as a model to study basic questions in genome biology. Using an interdisciplinary approach that combines biochemical, molecular, genetic, cellular, and computational techniques, we address how non-coding RNAs direct changes in 3D genome organization and determine gene expression states in health and disease. RNA has a special place in epigenetics by being uniquely capable of site-specific action (cis-regulation). Our recent discovery that "junk RNA" such as B2 SINEs can function as epigenetic ribozymes puts a new spin on the functions of long noncoding RNAs.
In recent years, we have also taken on the challenge of translating basic knowledge to treat X-linked intellectual disabilities, such as Rett Syndrome, Fragile X, and CDKL5 Syndrome. Our interests reside in the 4 main areas below. |

1. Xist RNA and the global transformation of the inactive X
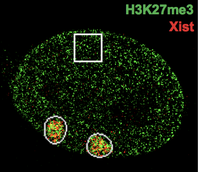
Xist RNA has served as an exemplary model to study the action of long noncoding RNAs in epigenetic regulation. How this RNA spreads along the X-chromosome and induces whole-chromosome silencing remains a major unsolved problem. Over the past decade, by developing and incorporating new RNA-centric methodologies, we have learned that Xist transforms the X-chromosome in 3 major ways: (i) It recruits and scaffolds silencing factors such as Polycomb repressive complexes to initiate inactivation (ii) In parallel, it repels activating factors such as the cohesins and SWI/SNF. And (iii) it transforms the Xi architecture into a unique 3D structure. Ongoing projects address the molecular underpinnings of each mechanism and include a deep dive into the roles of >100 protein partners identified by our recent Xist RNA proteomic analysis.
|

|
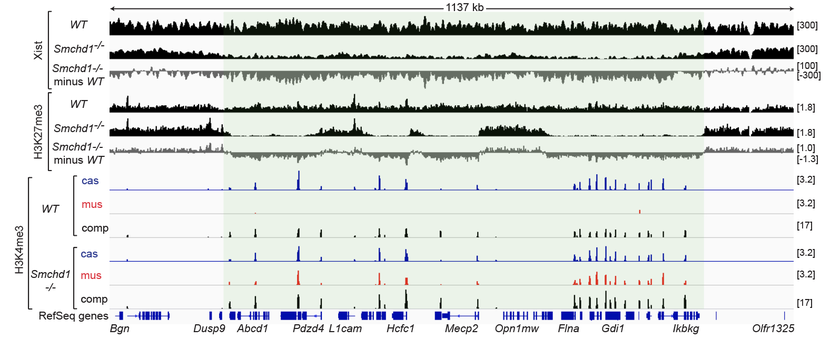
Wang CY, Jegu, T, Chu HP, Oh HJ, and Lee JT. (2018) SMCHD1 merges chromosome compartments and assists formation of Super-structures on the inactive X. Cell 174, 406-421.
SMCHD1 is required to spread Xist RNA, reshape the X-chromosome, and silence genes.
SMCHD1 is required to spread Xist RNA, reshape the X-chromosome, and silence genes.
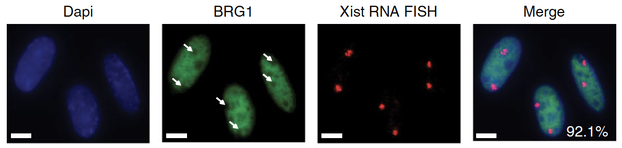
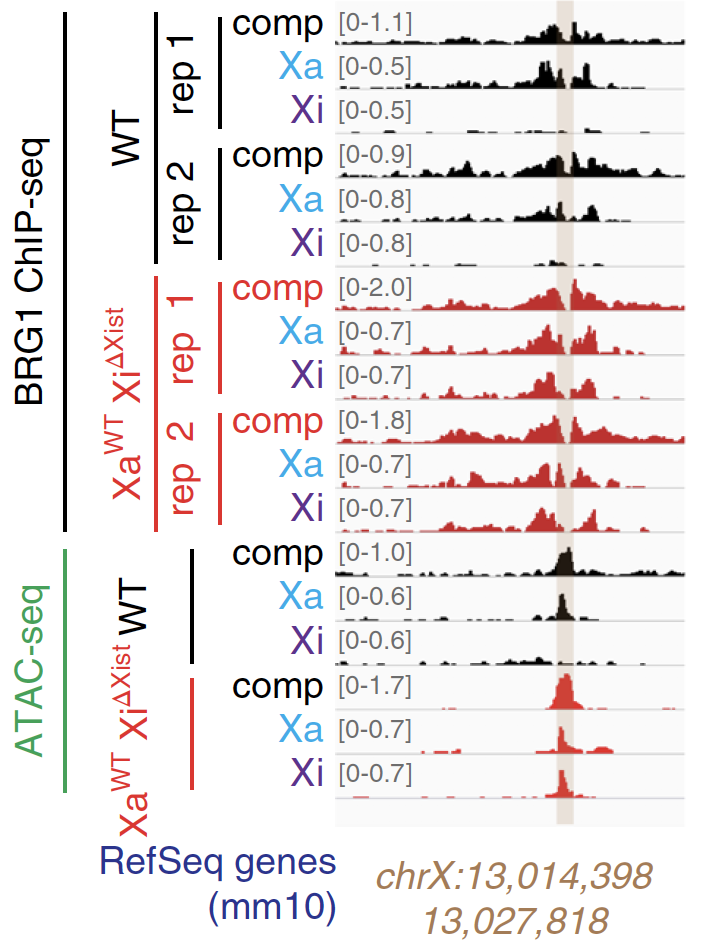
Jegu, T. †, Blum R. †, Cochraine, J.C., Yang, L., Wang, C.Y., Gilles, M.-E., Colognori, D., Szanto, A., Marr, S.K., Kingston, R.E., and Lee, J.T. (2019) Xist RNA antagonizes the SWI/SNF chromatin remodeler, BRG1, on the inactive X chromosome. Nat Struct Mol Biol 10.1038/s41594-018-0176-8.
Xist RNA repels this activating factor in order to suppress chromatin accessibility on the X chromosome. |
|
2. How the cells count X-chromosomes and choose just one for inactivation
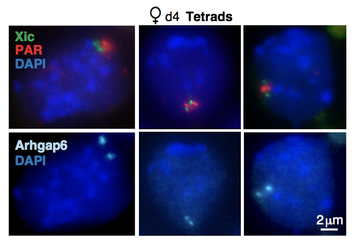
Chu HP, Froberg JE, Kesner B, Oh HJ, Ji F, Sadreyev R, Pinter SF, and Lee JT. 2017. PAR-TERRA directs homologous sex chromosome pairing. Nat Struct Mol Biol. 24,620-631.
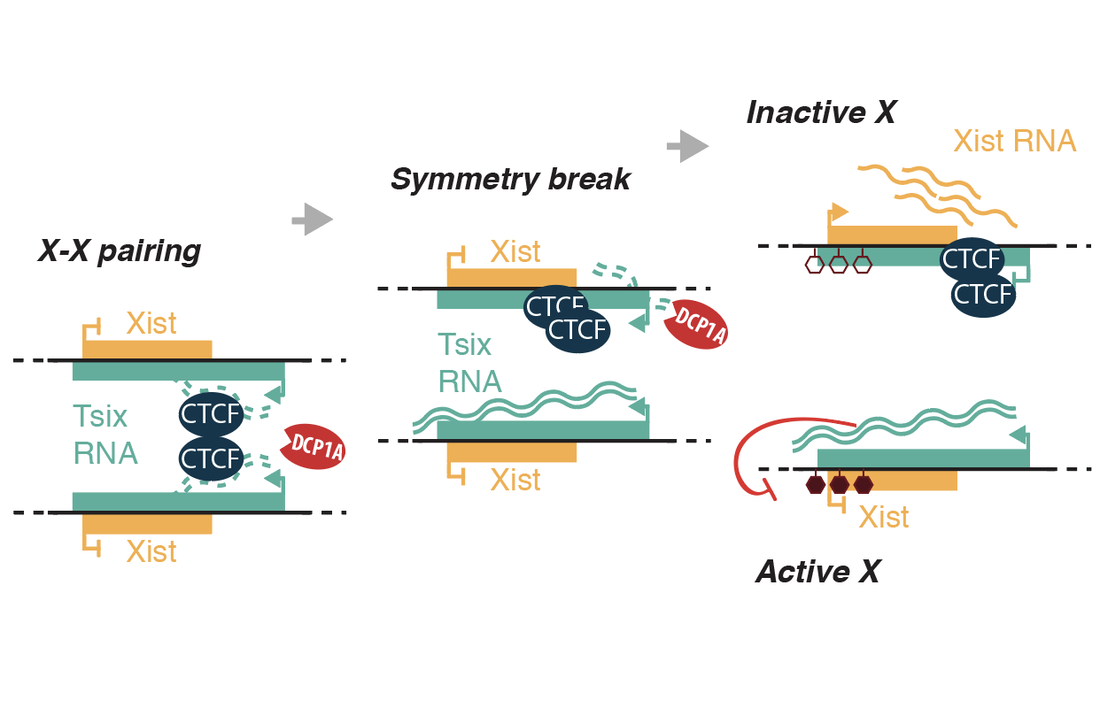
Aeby E, Lee H-G, Lee Y-W, Kriz A, del Rosario BC, Oh HJ, Boukhali M, Haas W, and Lee JT. 2020. Decapping enzyme 1A breaks X-chromosome symmetry by controlling Tsix elongation and RNA turnover. Nature Cell Biology, doi.org/10.1038/s41556-020-0558-0.
Sun S, Del Rosario BC, Szanto A, Ogawa Y, Jeon Y, and Lee JT. (2013). Jpx RNA activates Xist by evicting CTCF. Cell 153, 1537-1551.
|
TAG! You're it !!
X-chromosome PAIRING and the flipping of a bistable switch: In early development, each cell in the epiblast counts chromosomes and, when the X-to-autosome ratio (X:A) is 1.0 or greater, the cell undergoes X-chromosome inactivation (XCI). A random choice mechanism selects one of two X-chromosomes in the female cell for inactivation. We have been systematically dissecting the underlying mechanisms and have shown that three noncoding loci — Tsix, Xite, and X-linked telomeres — play key roles in both processes. Telomeric transcripts original from the pseudoautosomal region (PAR) that we call "PAR-TERRA" agglomerate telomeres and this telomeric agglomeration then drives the pairing of the two X-inactivation centers. The transient pairing event between two X-chromosomes requires Tsix and Xite, which in turn generate the epigenetic asymmetry that enables one X to become the Xi and the other to remain the Xa (active X). What does one X chromosome say to the other? Current projects seek to identify and characterize the molecular factors driving the epigenetic asymmetry. So far, we have identified one such factor by developing a new method of identifying RNA-protein interactions (BioRBP). This work shows that the RNA decapping enzyme, DCP1A, is a critical factor to break X-X symmetry and to enable CTCF to "flip" to one X chromosome — the future Xi — and enable Xist induction from that chromosome. How do cells count X chromosomes? Pairing is just one part of the counting-choosing conundrum. Recently, we have learned several other things about the counting process itself. X-linked and autosomal factors titrate each other to effectuate the X:A ratio, with the noncoding Jpx RNA serving as an X-linked “numerator” and CTCF serving as an autosomal “denominator”. Counting involves (in part) a titration between Jpx RNA and CTCF protein to measure the X:A ratio (see Video). Ongoing projects aim to understand how Jpx turns on Xist RNA and to identify additional counting elements. |
3. Mining the non-coding genome and its repetitive elements: Hidden ribozymes in B2 and ALU
|
Noncoding sequences make up 98% of our genome. Once thought to be nothing more than “junk”, the noncoding genome turns out to harbor many of the regulatory elements for the 2% of our DNA that encodes protein. About half of the noncoding space consists of “unique” sequence. The noncoding elements for X-chromosome inactivation reside in this unique noncoding space. In addition to the processes in 1 and 2 above, we are interested in how the noncoding sequences control genomic imprinting using imprinted X-inactivation as a model.
The other half of our noncoding genome consists of “repetitive elements”, including the retrotransposable elements that have come to dominate our noncoding space. Their functions largely remain unknown, but recent work from various labs, including our own, has started to elucidate their raison d’etre. For example, we have identified a surprising function for a specific type of repetitive element called the B2, a subclass of SINE retrotransposons. B2 RNA binds stress response genes in resting cells. Upon heat shock, the Polycomb protein, EZH2, is recruited to these genes, induces a site-specific cleavage of B2 RNA, and enables transcription of the stress response genes. This discovery suggests that the noncoding RNA from repetitive elements play a very important role in gene regulatory networks during stress. We would like to understand how B2 RNA binds to genes, how they block the advancement of RNA Polycomerase II, and how they are cleaved in the presence of EZH2.... And now we know that SINE retrotransposons, B2 and ALU, are SELF-CLEAVING RIBOZYMES! Their activity is enhanced 100-fold by contact with EZH2. Because of their sheer numbers in the mammalian genome, B2 and ALU are likely to represent the predominant ribozyme activity in mammals. Hernandez AJ, Zovoilis A, Cifuentes-Rojas C, Han L, Bujisic B, Lee JT. 2019. B2 and ALU retrotransposons are self-cleaving ribozymes whose activity is enhanced by EZH2. Proc Natl Acad Sci U S A. 2019 Dec 23. pii: 201917190. doi: 10.1073/pnas.1917190117. Read. |
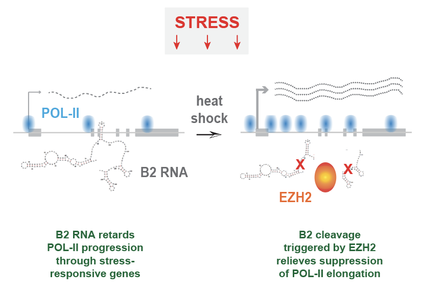
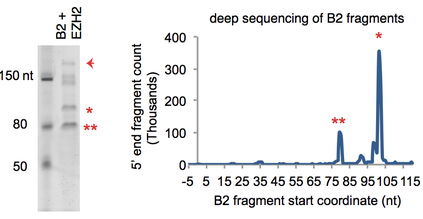
B2 RNA is cleaved in the presence of EZH2. Site-specific cleavage leads to activation of heat shock-responsive genes. Prior to heat shock, the binding of B2 RNA to these genes causes RNA Polymerase II to stall at promoter proximal regions, thereby regulating the rate of transcription across the stress-responsive genes. Zovoilis, A., Cifuentes-Rojas, C., Chu, H.P., Nernandez, A.J., and Lee, J.T. 2016. Destabilization of B2 RNA by EZH2 activates the stress response. Cell 167, 1788-1802.
|
4. Treating X-linked intellectual disabilities through X-reactivation
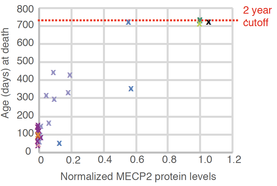
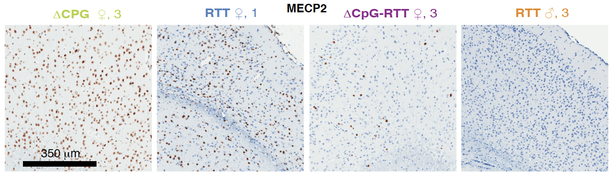
The inactive X (Xi) is a treasure trove of genes, but it lies dormant because it is subject to a robust silencing mechanism. For decades, it was thought that Xi reactivation is not possible once X-inactivation is established in somatic cells. However, recent work from our lab and others have shown that the Xi can be partially reactivated. This perturbability has led to the concept of treating X-linked disease in females by tapping into the normal but dormant allele on the Xi. Our research has led to new understanding of Xi reactivation, via both genetic and pharmacological means, and shown that Xist RNA is a key organizing principle for silencing as well as unsilencing. Disrupting Xist’s ability to recruit interacting proteins may hold the key to Xi-reactivation. Most exciting to us is a mixed modality approach that upregulates MECP2 protein by 30,000-fold — an unprecedented level that makes treating Rett Syndrome through Xi-reactivation a strong possibility to consider. We have also developed a new female mouse model for Rett Syndrome and are also developing models for other X-linked disorders. The ∆CpG-RTT female mouse develops a severe neuromotor disease and exhibits repetitive and self-injurious behaviors — two behaviors commonly seen in Rett Syndrome and other autistic disorders. Current efforts are directed at increasing the degree of MECP2 reactivation. The reactivation approach can potentially be applied to dozens of other X-linked disorders, including Fragile X and CDKL5 Syndromes. We currently have several preclinical candidates and are testing them in patient cells.
Read more in the Atlas of Science by clicking here.
Partial X-reactivation seems to be well-tolerated in a small animal model. Read more here.
We are also excited to share that our X-reactivation platform was selected as one of the "Disruptive Dozen" technologies at the 2022 World Medical Innovation Forum! (click here for video)
If interested in supporting our efforts in this space, please click below.
Read more in the Atlas of Science by clicking here.
Partial X-reactivation seems to be well-tolerated in a small animal model. Read more here.
We are also excited to share that our X-reactivation platform was selected as one of the "Disruptive Dozen" technologies at the 2022 World Medical Innovation Forum! (click here for video)
If interested in supporting our efforts in this space, please click below.
|
|
Left panel: Correlation between lifespan and MEPC2 protein levels in the brain of terminal stage mice. Right panel: MECP2 staining (brown) of fixed brain slices from different mouse genotypes as indicated. The ∆CpG-RTT mouse model shows very few MECP2+ brain cells, but survives significantly longer and performs better on neuromotor tests in a manner that correlates with its MECP2 expression level. From: Carrette L, Blum R, Ma W, Kelleher R, and Lee JT. 2018. A Tsix-Mecp2 female mouse model for Rett Syndrome reveals that low-level MECP2 expression extends life and improves neuromotor function. PNAS ePub July 23, 2018.
|
We are also developing new methods to reactivate FMR1 for Fragile X Syndrome. See our recent interview. |
|



